

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

求职招聘

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
求职招聘
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
正文
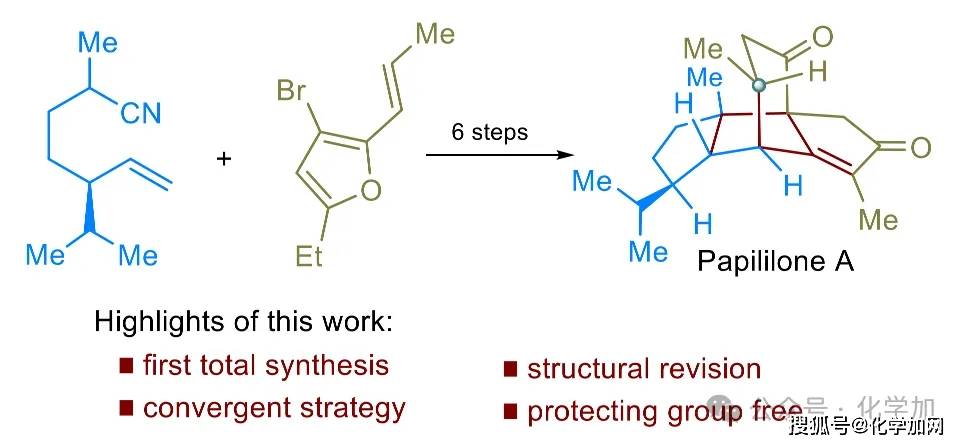
具有稠合桥环骨架和多立体中心的多环萜类分子(如calidoustene、pleuromutilin和papililone A)(图1a)因其复杂笼状结构和显著生物活性成为合成领域的研究热点。与pleuromutilin二萜所具有的5/6/8三环骨架不同,papililone A(1)拥有独特的5/6/5/5稠合桥环四环骨架和相邻的季碳立体中心,显著提升了分子复杂性。这些分子的精巧结构不断激发合成化学家探索新型反应,如自由基环化、环化/重排串联反应等,以实现复杂天然产物及其类似物的高效、简洁合成。尽管近年来已取得一定进展,但开发普适性强、效率高的方法,以快速构建拥挤的笼状多环体系,仍是当前合成化学领域亟待解决的关键挑战。
Papililone A(1)是由贵州大学周康课题组于2022年从真菌Papiliomyces sp.中分离获得的一种新型二萜类天然产物,具有新颖的四环骨架和良好的抗炎活性(图1a)。从合成角度来看,其空间拥挤的笼状多环骨架带来了多重挑战:(a)稠合的线性三奎烷结构(A-C环)与高张力的六元桥环(D环)紧密连接,形成高度刚性的笼状骨架;(b)分子中包含六个连续的立体中心,其中包括两个相邻且高度拥挤的桥头全碳季碳中心,以及一个大位阻的β-异丙基取代基,增加了化学合成的难度。此外,近期的计算研究指出,该天然产物的C17-甲基构型应从先前推测的α构型修正为β构型,这也进一步提升了其合成的复杂性。截至目前,papililone A的全合成尚未见报道。
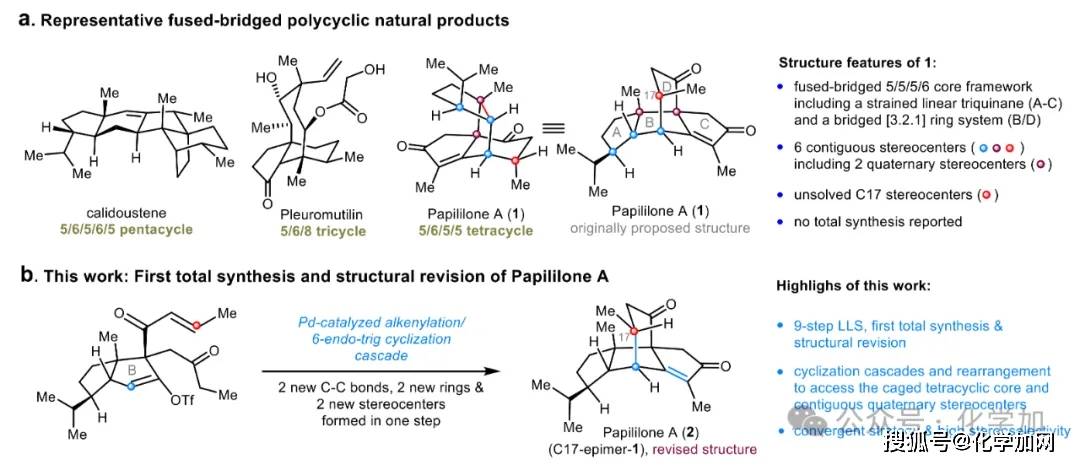
图1. 代表性稠合并桥多环萜类分子及Papililone A的首次全合成
近日,侯四化课题组利用极性-自由基串联环化、立体选择性1,2-加成、插烯α-醇酮重排、钯催化烯基化/共轭加成串联环化等关键反应,以9步线性最长步骤和无保护基的合成策略实现了(-)-papililone A的首次全合成,并修正了其C17位的立体构型。
在逆合成分析中(图2),作者提出天然产物papililone A(1)及其C17差向异构体(2)可通过双烯醇负离子过渡态TS-1经钯催化的酮烯基化/6-endo-trig串联环化反应,由化合物3高效构建。该串联反应可以一步形成两个新的C–C键、两个新的立体中心(C6和C17),并构建目标分子中关键的CD环系。中间体3的合成则依赖于关键的插烯α-醇酮重排反应与三氟甲磺酰化反应,起始原料为α,β-不饱和酮化合物4。该结构重组策略巧妙利用前体4中C19=O与C7–OH之间的分子内氢键(C19=O···HO–C7),有效调控反应构象,从而精准控制C1→C11的迁移过程,实现C11位全碳季碳立体中心的立体选择性构建。化合物4可通过汇聚式偶联策略制备:以呋喃片段5与环丁酮6进行1,2-加成,随后经氧化反应得到。而化合物6则可由腈基前体7经优势反应构象7a的非对映选择性极性-自由基环化反应高效构建。
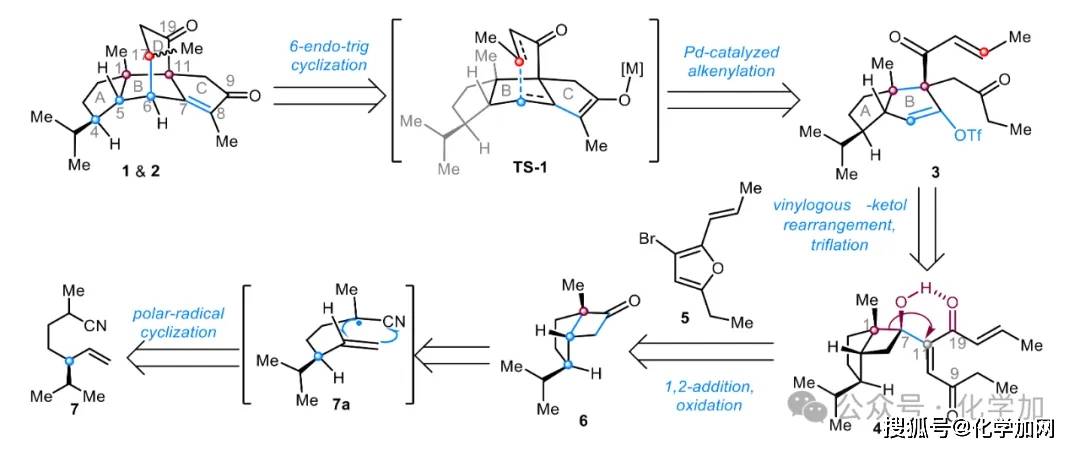
图2. Papililone A的逆合成分析
如图3所示,片段5和6的合成路线如下:已知炔丙醇8(一步制备)在i-PrMgCl存在下与Weinreb酰胺发生炔基化反应,以79%的收率得到炔酮9。化合物9在Obrecht条件下(HBr/HOAc)发生溴化环化反应,以54%的收率、克级规模制备3-溴呋喃5。烷基溴10(一步制备)与甲基丙烯酸酯在Mg、CuCl和TMSCl条件下发生Michael加成,经水解后以53%的收率得到羧酸11。最初,作者尝试通过酰胺12(由11与吡咯烷缩合制得)的 [2+2] 环化反应构建环丁酮6。尽管该反应收率较高,但非对映选择性较差(d.r. = 1:1.5),且主要生成不需要的非对映异构体6'。为提高立体选择性,作者转而采用Fleming极性-自由基环化策略,以期实现[2+2]环化的立体控制。经优化反应条件,ω-烯基腈7(由11与NH4HCO3反应制得)成功以60%的收率转化为单一非对映异构体6,非对映选择性高达d.r. > 20:1。该优异的立体选择性可能是由于β-异丙基取代基的空间位阻控制。化合物6的结构及相对构型通过其衍生物6a的X射线单晶衍射分析得以确证。
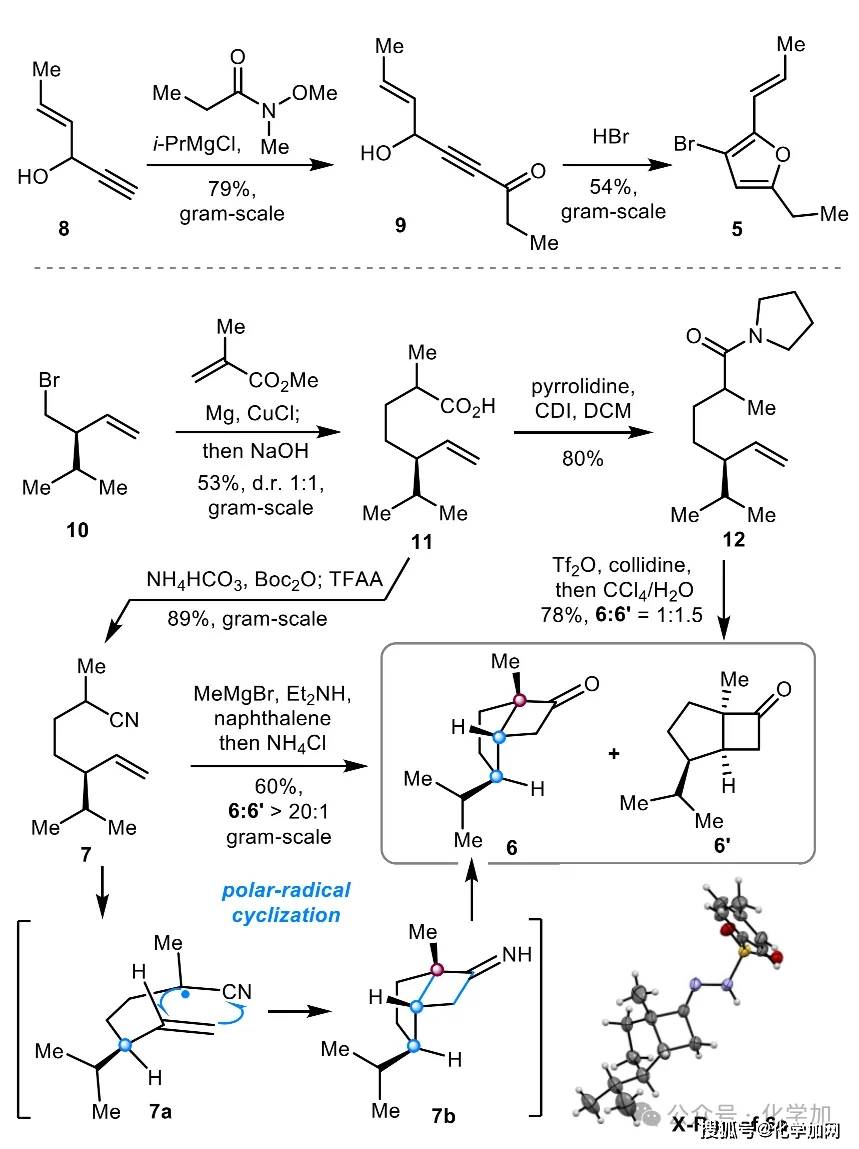
图3. 片段5和6的制备
如图4所示,3-溴呋喃5经n-BuLi处理发生锂-卤交换后,与5/4并环环丁酮6反应,以79%的收率得到非对映异构体混合物13/13'(d.r. = 1.1:1)。值得注意的是,引入镧盐LaCl₃·2LiCl作为添加剂后,反应的非对映选择性显著提升,以77%的收率获得单一非对映体13(d.r. > 20:1)。该反应的高立体选择性可能归因于LaCl₃·2LiCl与酮羰基在空间位阻较小的凸面螯合,从而迫使亲核试剂从更易接近的凹面进攻,从而实现立体控制。随后,在氧气、亚甲蓝和光照条件下,13/13'中的呋喃结构单元发生化学选择性氧化裂解,分别以87%和56%的收率转化为重排前体4和4',为后续的重排反应提供关键中间体。
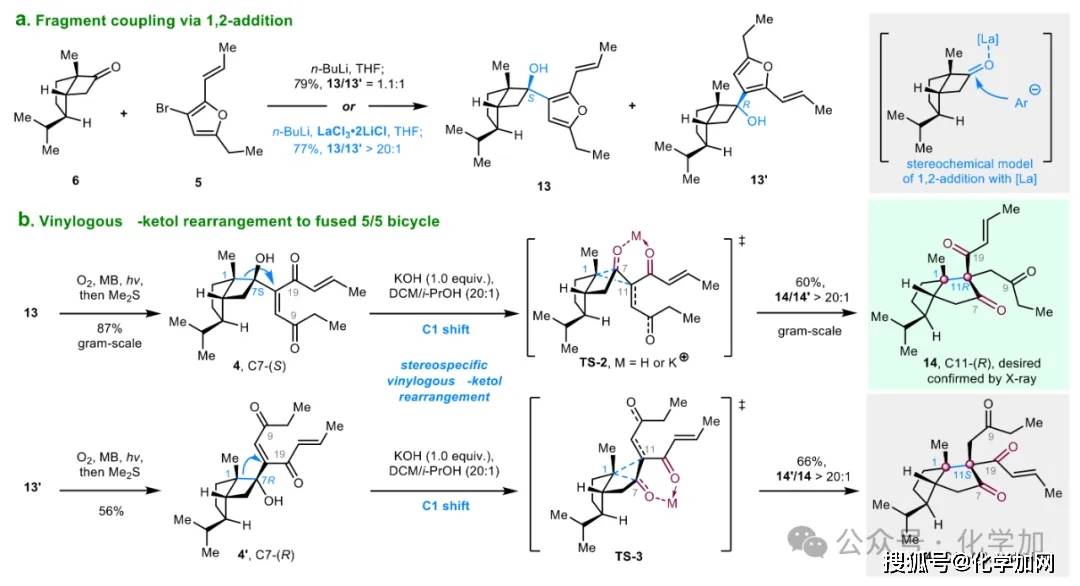
图4. 汇聚式策略合成5/5并环化合物
作者随后对关键的插烯α-醇酮重排反应进行了系统研究。尽管该重排反应已应用于α,β-不饱和环酮体系,但其在含不稳定共轭二烯酮的线性α,β-不饱和酮体系(如4)中的研究较少。初步尝试在路易斯酸(EtAlCl₂、BF₃·Et₂O、TMSOTf)条件下,底物4虽能完全消耗,但未能生成目标产物14。随后,作者发现KOH/i-PrOH/DCM体系可有效促进该重排反应,以60%收率得到预期目标产物14(d.r. > 20:1),其结构及新形成的C11位季碳的构型(11R)通过X射线单晶衍射得以确证。DFT计算表明,该重排过程的高立体选择性源于分子内氢键(C19=O···HO–C7)与空间位阻的协同调控,使反应通过能量更低的过渡态TS-2进行,从而控制重排反应的面选择性。相应地,前体4'(C7差向异构体)在相同条件下以66%收率(d.r. > 20:1)转化为产物14'(C11差向异构体),其C11季碳构型翻转为11S,进一步验证了该重排反应的立体化学机制的可靠性。
最后,作者尝试传统羰基缩合的合成策略,以aldol或Michael反应为关键步骤,构筑papililone A的高张力5/5/6三环结构(B-D环)(图5)。然而,在不同酸性(PTSA、(PhO)₂PO₂H)和碱性条件(HTMP、NaH、KHMDS)下,预期的aldol或Michael反应均未发生,无法构建目标分子的多环骨架。令人高兴的是,在LiHMDS与Comins试剂的反应条件下,三羰基化合物14以64%的收率转化为烯基三氟甲磺酸酯3,成为该合成路线的关键突破口。基于此,作者转而采用过渡金属催化策略,即通过钯催化的酮烯基化/6-endo-trig环化反应构建CD环,突破现有的合成瓶颈。尽管烯醇负离子的6-endo-trig环化(如中间体Int-1)因不利的立体电子效应而极具挑战,但系统优化发现钯催化剂对反应路径具有决定性影响:当使用PdCl₂或Pd(TFA)₂与配体DIOP时,反应主要发生烯基化/质子化过程,生成aldol型产物15(表1,条目1–2);而采用Pd(MeCN)₂Cl₂时,可有效促进烯基化/6-endo-trig环化串联反应,以13%收率得到笼状四环产物2(d.r. > 20:1)(条目3)。该产物的结构与立体化学经X射线单晶衍射得以确证。进一步降低配体用量可提升产物2的收率(条目4–5)。最终,在优化条件 [Pd(MeCN)2Cl2 (10 mol%)、DPPP (8 mol%)、甲苯、140°C] 下,以45%的收率获得2(d.r. > 20:1)。DFT计算表明,6-endo-trig环化的高非对映选择性源于椅式过渡态TS-1比船式过渡态TS-4低3.9 kcal/mol。
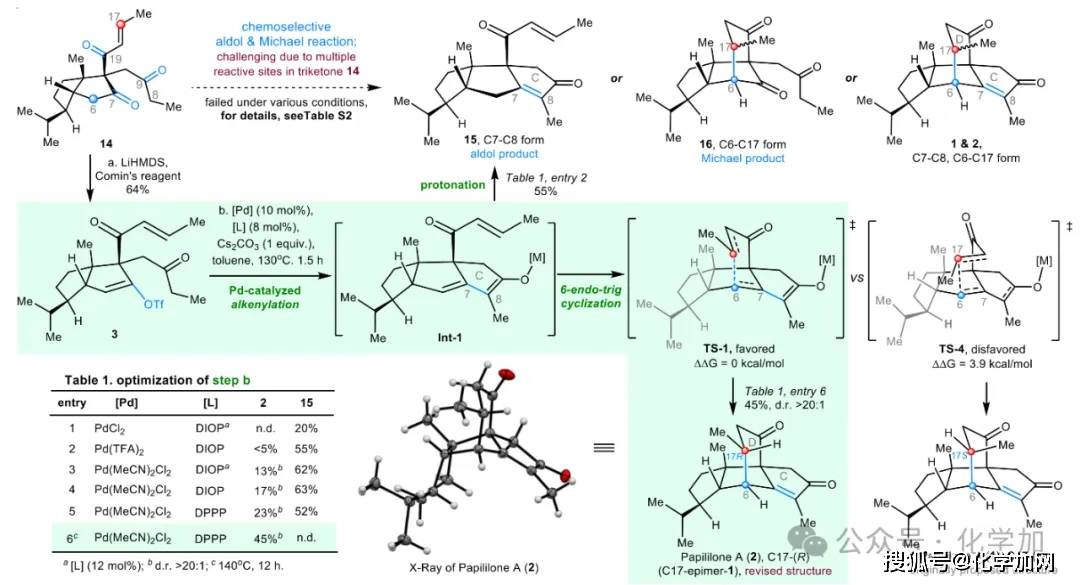
图5. 天然产物papililone A的全合成
化合物2的核磁共振谱图数据与天然papililone A完全一致,证实天然产物papililone A的真实结构为C17-(R)构型的2,而非最初所指定的C17-(S)构型。基于上述合成路线,由 (+)-10制备的(–)-papililone A(2)的圆二色谱(ECD)和旋光度均与天然样品一致,从而确证了天然(–)-papililone A的绝对构型。
总结
侯四化团队的这项工作不仅实现了papililone A的首例全合成,更展示了一种简洁、高效的合成策略。该策略的核心在于巧妙融合多种精心设计的新型反应:通过钯催化的烯基化/6-endo-trig环化串联反应构建5/5/6三环骨架;借助螯合导向的插烯α-醇酮重排精准控制C11立体中心;并利用极性-自由基串联环化构建环丁酮结构,有效调控环间连接的立体化学。此外,作者还巧妙地以呋喃前体为关键中间体,有效控制了共轭双烯酮中双键的几何构型,为后续立体选择性重排反应奠定了基础。这一系列反应的协同应用,充分体现了合成设计的逻辑性与创新性。该合成不仅成功解决了papililone A的结构争议,确立了其正确的构型,还为复杂多环萜类化合物的合成提供了新颖的策略与方法。这一成果也为后续相关生物活性研究和药物开发奠定了坚实的基础。
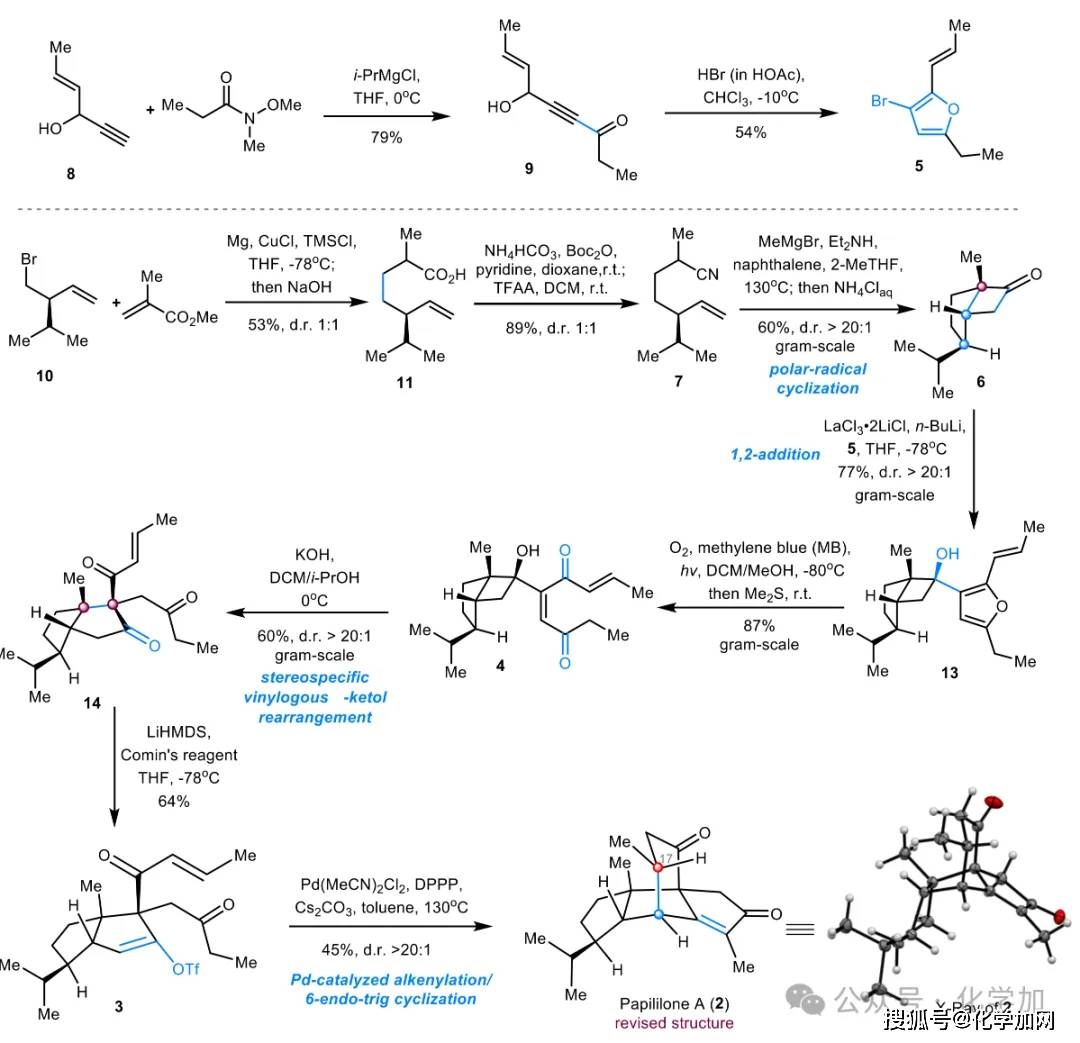
图6. 天然产物papililone A的合成路线
上海交通大学药学院2023级博士研究生单兴钱为论文第一作者,侯四化长聘教轨副教授为论文通讯作者。
文献详情:
Xing-Qian Shan, Xiang Zhang, Peng-Fei Zheng, Bao-Kuan Guo, Yong-Qiang Tu, Si-Hua Hou*
Convergent Total Synthesis of Papililone A via Pd-Catalyzed Alkenylation/Cyclization Cascade.
J. Am. Chem. Soc. 2025, 10.1021/jacs.5c17278
https://doi.org/10.1021/jacs.5c17278
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
